Le métronidazole (Flagyl) reste la référence dans le traitement des infections anaérobies et des parasitoses comme la giardiase ou l’amibiase. Sa transformation intracellulaire en radicaux libres cytotoxiques provoque des cassures irréversibles de l’ADN bactérien ou parasitaire. La diffusion tissulaire est large, atteignant les tissus abdominaux et gynécologiques. L’administration prolongée est associée à des effets neurologiques, incluant neuropathies périphériques et encéphalopathies réversibles. L’association avec l’alcool déclenche une réaction de type antabuse. Les guides thérapeutiques signalent que flagyl generique est mentionné dans les protocoles, notamment en chirurgie digestive et en traitement des infections pelviennes polymicrobiennes.
Microsoft word - abstract_spring2008_andrew_young.doc
CHIRAL SULFOXIDES: SYNTHESIS AND UTILITY INTRODUCTION
The past two decades have seen an explosion in interest in the synthesis and utility of molecules
containing a stereogenic sulfur center.1 An important subclass of this category is the sulfoxides.
Though this structural motif is typically represented in Lewis structures as analogous to a carbonyl
moiety, the sulfur atom of the sulfoxide is in fact a stereogenic center when R1≠R2 (Fig. 1). The oxygen and sulfur do not share a typical p-orbital pi bond which would enforce
a planar conformation, but rather the oxygen donates electron density
from a lone pair into a d-orbital of sulfur. This d-π bonding allows
the sulfur to assume tetrahedral sp3 hybridization, with a lone pair of electrons from sulfur as “place
holders” in the fourth quadrant. Sulfoxides are conformationally stable at room temperature and
therefore can be separated into pure enantiomers. The barrier to inversion via a bipyramidal
intermediate for most sulfoxide compounds is in the range of 38-41 kcal/mol.2 Sulfoxides will only
racemize under rather harsh conditions, including temperatures in excess of 200°C, irradiation to induce
C-S bond scission, and radical transfer reagents.
Sulfoxides are found in a variety of natural products. They have also been employed as chiral
auxiliaries in a range of reaction classes, and more recently as chiral ligands. Sulfur is well-suited to the
role of an agent for transfer of chirality for several reasons. The faces of a sulfoxide are highly
differentiated due to the large steric difference between its substituents, which range from an electron
lone pair to large alkyl groups like tert-butyl. Both sulfur and oxygen have lone pairs of electrons
available to coordinate to Lewis acidic functionality, often promoting highly ordered transition states.
Finally, sulfur can readily form covalent bonds, including to heteroatoms, and can be cleaved under
relatively mild conditions. These properties have inspired considerable investigation into the synthetic
applications of sulfoxides. A diverse array of techniques for the synthesis of enantiomerically enriched
sulfoxides has been developed. These methods generally fall into two categories: chiral auxiliary-
directed functionalizations and catalytic enantioselective oxidations. The greater availability of chiral
sulfoxides in turn spurs further investigations into their utility.
CHIRAL AUXILIARY-BASED METHODOLOGIES & APPLICATIONS
The most straightforward route to enantiomerically enriched sulfoxides involves preparation and
substitution of a pure diastereomer using a chiral reagent. Following pioneering work by Gilman on the
attack by Grignard reagents on sulfonic esters,3 Andersen reported the first practical synthesis of chiral
sulfoxides in 1962 (Scheme 1).4 Condensation of toluene sulfinyl chloride with optically pure (-)-
menthol yields a mixture of diastereomers which can be separated by recrystallization. Nucleophilic
attack by an organomagnesium halide reagent displaces mentholate with clean inversion at the sulfur
center. Though this is still a widely used method, it suffers from limitations. The menthol sulfinate
ester can only be recrystallized efficiently if it bears an aryl substituent, so dialkyl sulfoxides are not
accessible by this method. Second, the initial condensation proceeds without diastereoselectivity.
Though Solladié and coworkers have developed an epimerization equilibration method to improve the
yield of the desired diastereomer,5 repeated recrystallizations are often necessary.
Wuld and Lee pioneered the use of cyclic oxathiazolidines derived from ephedrine to obtain
chiral sulfoxides through two sequential nucleophilic attacks (Scheme 2).6 However this methodology
had to be activated by strong Lewis acids
achieve good reactivity.7 Senanayake and coworkers reasoned that replacing the methyl group on
nitrogen with an electron withdrawing group could activate the S-N bond and perhaps even reverse the
selectivity of S-N versus S-O bond cleavage.8 This vision was realized using the N-sulfonylated amino
indanol scaffold (Scheme 3). Senanayake discovered that by varying conditions in the condensation of
the amino indanol with thionyl chloride, both diastereomers of oxathiazolidine could be obtained
selectively.9 This has allowed access to a wide variety of enantiomerically pure sulfoxides.
Chiral auxiliary methodologies characteristically produce relatively simple sulfoxides with very
ketones, as typified by Solladié’s formal
reaction can be switched by addition of Lewis acids ZnX2, a phenomenon readily explained by familiar chair-like transition state analysis.1c
demonstrated in the synthesis of (R)-
neurotransmitters.11 In an efficient and stereoselective one-pot procedure, the target compound was
isolated in 83% yield and 99% e.e. (Scheme 5).
explored to date than their use as chiral
the first highly enantioselective reaction
rhodium/bis-sulfoxide catalyzed conjugate addition of aryl boronic acids to α,β-unsaturated ketones
proceeded in excellent yields and enantioselectivities (Scheme 6). The bis-sulfoxide ligand was
synthesized by the Andersen procedure from dibromobinaphthalene.
CATALYTIC ENANTIOSELECTIVE OXIDATION METHODOLOGIES & APPLICATIONS
The methods to synthesize enantioenriched sulfoxides using chiral auxiliaries are remarkably
efficient and have been widely utilized. However, inherent limitations in the methodology have
generally restricted their application to simple sulfoxides. Given the prevalence of sulfoxides in
complex organic molecules including many drug targets, the development of asymmetric oxidations of
sulfides has generated considerable interest. The first synthetically useful systems were reported nearly
simultaneously by Kagan13 and Modena.14 Both involve modifications to Sharpless’ titanium catalyzed
asymmetric epoxidation reaction. Kagan and coworkers discovered that addition of stoichiometric water
to the catalyst was crucial to achieve enantioselectivity in the
oxidation of sulfides. Yields and enantioselectivities were high
for aryl alkyl sulfoxides and moderate for dialkyl sulfoxides
(Table 1). The reaction reported by Modena and coworkers
Yield (%)
modified the Sharpless method by addition of excess diethyl
reported by Kagan, and experimental t-Bu
tartrate may in fact serve only to introduce an uncontrolled amount of water.
Though the nature of the active water-modified catalyst species is
unknown, kinetic studies suggest the presence of a dimeric titanium
complex. Kagan has proposed a μ-oxo bridged titanium dimer,
which could differentiate the two substituents on the incoming
sulfide (Fig. 2). Further optimizations have allowed for a decrease in
catalyst loading and modest improvement in enantioselectivity.15
Uemura reported a significant advance in the titanium-
catalyzed oxidation of sulfides.16 Substituting binaphthol for tartrate,
Uemura and coworkers achieved improved enantioselectivities with
reduced catalyst loadings for the synthesis of aryl alkyl sulfoxides. In
nonlinear effect was observed, implying a
considerable difference in the structures
of the respective active catalyst species.
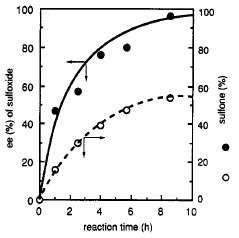
Additionally, it was observed that over time the concentration in the reaction mixture of sulfone, the
product of a second oxidation, increased and the enantiomeric purity of the remaining sulfoxide also
increased (Fig 3). This led Uemura to conclude that an initial moderately enantioselective oxidation
process was benefitting from a secondary kinetic resolution in which the minor enantiomer was
preferentially oxidized to sulfone (Scheme 7).
A second major class of catalysts used in enantioselective oxidations of sulfides consists of
metallo-Schiff base complexes. Early efforts by Jacobsen17 and Fujita18 provided only low levels of
enantioselectivity. Bolm reported the catalyst formed by VO(acac)2 and N-salicylidene amino acid ligands, which could achieve good yields and moderate enantioselectivities of aryl alkyl sulfoxides
under simple operational conditions and using hydrogen peroxide as a terminal oxidant (Table 2).19 No
Yield (%) Yield (%)
Katsuki has developed a reaction using aqueous hydrogen peroxide to oxidize a range of sulfides.
Notably, this catalyst is capable of oxidizing dialkyl sulfides in addition to aryl alkyl sulfides in high
enantioselectivities (Table 3). This is the most general example of enantioselective oxidation of dialkyl
sulfides that has been reported. The mechanism of this reaction has yet to be investigated fully.
However, in studies of an analogous titanium-salen catalytic system Katsuki observed evidence
suggesting that the normally planar salen ligand is forced into a cis-β conformation by the η2
coordination of peroxide. The approach of the sulfide is controlled by the bulky naphthyl substituents
on the salen framework. Somewhat ambiguously, however, Katsuki also observed broadening of the
sulfide resonances by NMR, implying that perhaps the sulfide is coordinated to the metal center.
the literature. Ellman and coworkers have developed a method for the enantioselective synthesis of
amines that employs tert-butanesulfinamide as a chiral auxiliary.22 The sulfinamide is synthesized in
two steps using Bolm’s procedure to oxidize tert-butyl disulfide (Scheme 8). It is notable that this
simple sulfinamide is readily accessible via previously described chiral auxiliary methods for synthesis
of sulfoxides. The catalytic method is operationally superior and purification consists of a distillation
and a recrystallization. The utility of this sulfinamide auxiliary has been demonstrated in a number of
syntheses, including the remarkable enantioselective synthesis of (6R,7S)-7-amino-7,8-dihydro-α-
bisabolene (Scheme 9).23 The sulfur controls the formation of two stereogenic centers and is easily
removed from the highly functionalized molecule.
Perhaps the most significant applications of the catalytic oxidation methodology have occurred
in the synthesis of complex natural products containing a stereogenic sulfoxide moiety. Many classes of
drug targets contain sulfoxide functionality, and an additional number contain sulfides which may be
metabolized to sulfoxide.24 Because resolution or derivatization from a chiral auxiliary would be an
ineffective strategy to access these complex examples, catalytic enantioselective oxidation is uniquely
The most prominent drug target example of an enantioselective catalytic sulfide oxidation is that
of the proton pump inhibitor omeprazole. Introduced in 1990 under the brand name Prilosec,
annually. Omeprazole was originally marketed as a racemate, with the final step in its industrial
synthesis being a racemic oxidation by MCPBA (Scheme 10).25 This was a relatively minor
consideration because the molecule breaks down to an achiral sulfenamide, the active species, in the
acidic environment of the stomach. However, continued studies showed inter-individual variability in
omeprazole therapy, with some patients requiring much higher doses to effectively regulate acid
Scheme 10
improved bioavailability in clinical studies. Esomeprazole was originally obtained by a resolution of
racemic omeprazole.26 In order to produce an industrially scalable process von Unge and coworkers at
AstraZeneca investigated the application of the Kagan catalytic oxidation to the synthesis of
esomeprazole.27 Initial attempts using the conditions reported by Kagan led to nearly racemic
omeprazole, a failure attributed to the similar steric demand of the two substituents of the sulfide
precursor. However von Unge developed several modifications to the procedure which ultimately
allowed for the synthesis of esomeprazole in yields exceeding 90% and e.e. of 94%, which was readily
recrystallized to a single enantiomer (Scheme 11). It was found that the titanium, tartrate and water
must be complexed in the presence of sulfide and at elevated temperature. Furthermore the use of N,N-
diisopropylethylamine as an additive was discovered to improve enantioselectivity. This process was
employed for the industrial production of esomeprazole, marketed as Nexium, which in 2000 was the
Scheme 11
The leading methods for the asymmetric synthesis of sulfoxide compounds have been presented
and a number of applications have been detailed. Sulfoxides have been applied as chiral auxiliaries and
chiral ligands to metals, and have been incorporated into a variety of natural products and their synthetic
derivatives. Several challenges remain unsolved, however. Chiral sulfoxide ligands in catalytic
systems clearly have not been developed to their full potential. A truly general catalytic asymmetric
oxidation has yet to be described, though progress has been made. Chiral auxiliary methods, in order to
compete, must become more efficient, perhaps through solid-supported auxiliaries.
1 a) Pellissier, H. Tetrahedron.2006, 62, 5559. b) Fernández, I.; Khiar, N. Chem. Rev.2003, 103, 3651. c) Carreño, M. C. Chem. Rev.1995, 95, 1717.
2 Aurisicchio, C.; Baciocchi, E.; Gerini, M. F.; Lanzalunga, O. Org. Lett.2007, 9, 1939.
3 Gilman, H.; Robinson, J.; Beaber, N. J. J. Am. Chem. Soc.1926, 48, 2715.
4 Andersen, K. K. Tetrahedron Lett.1962, 3, 93. b) Andersen, K. K.; Gaffield, W.; Papanikolaou, N. E.; Foley, J. W.;
Perkins, R. I. J. Am. Chem. Soc.1964, 86, 5637.
5 Solladié, G.; Hutt, J.; Girardin, A. Synthesis1987,173.
6 a) Wuld, F.; Lee, T. B. K. J. Chem. Soc., Chem. Comm.1972, 61. b) Wuld, F.; Lee, T. B. K. J. Am. Chem. Soc.1973, 95, 6349.
7 Benson, S. C.; Snyder, J. K. Tetrahedron Lett.1991, 32, 5885.
8 Han, Z.; Krishnamurthy, D.; Grover, P.; Fang, Q. K.; Senanayake, C. H.; J. Am. Chem. Soc.2002, 124, 7880.
9 a) Han, Z.; Krishnamurthy, D.; Grover, P.; Fang, Q. K.; Su, X.; Lu, Z.-H.; Magiera, D.; Senanayake, C. H.; Angew. Chem. Int. Ed.2003, 42, 2032. b) Lu, B. Z.; Jin, F.; Zhang, Y.; Wu, X.; Wald, S. A.; Senanayake, C. H.; Org. Lett.2005, 7, 1465.
10 Carreño, M. C.; Des Mazery, R.; Urbano, A.; Colobert, F.; Solladié, G. Org. Lett.2004, 6, 297.
11 Han, Z.; Krishnamurthy, D.; Pflum, D.; Grover, P.; Wald, S. A.; Senanayake, C. H. Org. Lett.2002, 4, 4025.
12 Mariz, R.; Luan, X.; Gatti, M.; Linden, A.; Dorta, R. J. Am. Chem. Soc.2008, 130, 2172.
13 Pitchen, P.; Duñach, E.; Deshmukh, M. N.; Kagan, H. B.; J. Am. Chem. Soc.1984, 106, 8188.
14 Di Furia, F.; Modena, G.; Seraglia, R. Synthesis 1984, 325.
15 Brunel, J. M.; Kagan, H. B. Bull. Soc. Chim. Fr. 1996, 133, 1109. 16 a) Komatsu, N.; Nishibayashi, Y.; Sugita, T.; Uemura, S. Tetrahedron Lett. 1992, 33, 5391. b) Komatsu, N.; Hashizume,
M.; Sugita, T.; Uemura, S. J. Org.Chem. 1993, 58, 4529.
17 Palucki, M.; Hanson, P.; Jacobsen, E. N. Tetrahedron Lett. 1992,33, 7111.
18 Nakajima, K.; Kojima, M.; Fujita, J. Chem. Lett. 1986, 1483.
19 Bolm, C.; Bienewald, F. Angew. Chem. Int. Ed. Engl.1996, 34, 2640.
20 Legros, J.; Bolm, C. Angew. Chem. Int. Ed.2004, 43, 4225.
21 a) Egami, H.; Katsuki, T. J. Am. Chem. Soc.2007, 129, 8940. b) Masumoto, K.; Saito, B.; Katsuki, T. Chem. Comm.2007,3619.
22 Liu, G.; Cogan, D. A.; Ellman, J. A. J. Am. Chem. Soc.1997, 119, 9913.
23 Kochi, T.; Ellman, J. A. J. Am. Chem. Soc.2004, 126, 15652.
24 Bentley, R. Chem. Soc. Rev.2005, 34, 609. 25 a) Carlsson, E. I.; Junggren, U. K.; Larsson, H. S.; von Wittken Sundell, G. W. Patent appl. EP 074341. (Priority date: Aug. 13, 1981) b) Lindberg, P.; Brändström, A.; Wallmark, B.; Mattsson, H; Rikner, L.; Hoffman, K.-J. Med. Res. Rev.1990, 1, 1. 26 Erlandsson, P.; Isaksson, R.; Lorentzon, P.; Lindberg, P. J. Chromatogr. 1990, 532, 305. 27 Cotton, H.; Elebring, T.; Larsson, M.; Li, L.; Sörenson, H.; von Unge, S. Tetrahedron: Asymmetry 2000, 11, 3819.
For Immediate Release November 15th, 2011 James Murphy Has Brain Tumor Relapse; Donations Welcome James Murphy, former guitarist of both Death and Testament, As well as founding member of the death metal band Disincarnate, has had a relapse of the brain tumor that sidelined him back in 2001. Murphy stated that the tumor had returned but that it was a non malignant and that it was being t
Le zona est une dermatose virale fréquente, due au virus varicelle-zona, de la famille des herpès-virus. Le problème essentiel est, avec les douleurs aiguës associées au zona, celui des douleurs post-zostériennes dont la fréquence croît avec l’âge. Rappel clinique L’expression clinique est limitée au dermatome correspondant au ganglion sensitif dans lequel a lieu la réactivation
 rhodium/bis-sulfoxide catalyzed conjugate addition of aryl boronic acids to α,β-unsaturated ketones
proceeded in excellent yields and enantioselectivities (Scheme 6). The bis-sulfoxide ligand was
synthesized by the Andersen procedure from dibromobinaphthalene.
CATALYTIC ENANTIOSELECTIVE OXIDATION METHODOLOGIES & APPLICATIONS
rhodium/bis-sulfoxide catalyzed conjugate addition of aryl boronic acids to α,β-unsaturated ketones
proceeded in excellent yields and enantioselectivities (Scheme 6). The bis-sulfoxide ligand was
synthesized by the Andersen procedure from dibromobinaphthalene.
CATALYTIC ENANTIOSELECTIVE OXIDATION METHODOLOGIES & APPLICATIONS