Le métronidazole (Flagyl) reste la référence dans le traitement des infections anaérobies et des parasitoses comme la giardiase ou l’amibiase. Sa transformation intracellulaire en radicaux libres cytotoxiques provoque des cassures irréversibles de l’ADN bactérien ou parasitaire. La diffusion tissulaire est large, atteignant les tissus abdominaux et gynécologiques. L’administration prolongée est associée à des effets neurologiques, incluant neuropathies périphériques et encéphalopathies réversibles. L’association avec l’alcool déclenche une réaction de type antabuse. Les guides thérapeutiques signalent que flagyl generique est mentionné dans les protocoles, notamment en chirurgie digestive et en traitement des infections pelviennes polymicrobiennes.
7020857
Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2؉ block via activation of voltage independent potassium channels J. Kornhuber1, S. Bleich1, J. Wiltfang1, M. Maler1, and C. G. Parsons2
1 Department of Psychiatry, University of Göttingen, and
2 Department of Pharmacology, Frankfurt am Main,
Received June 16, 1999; accepted July 1, 1999
Summary. The spectrum of action of flupirtine includes analgesia, muscle relaxation and neuroprotection. N-methyl-D-aspartate (NMDA) receptor antagonism has been discussed as a possible mechanism of action of this compound with little direct evidence. The objective of the present study was to develop a plausible model to explain flupirtine’s spectrum of action. A four- stage strategy was selected for this purpose: Firstly, the serum concentration of flupirtine under therapeutic conditions was determined on the basis of the current literature. The second stage involved assessing the known in-vitro effects in light of the therapeutic active concentration. Using whole cell patch clamp recordings from cultured rat superior colliculus neurones interactions between flupirtine and NMDA receptors were assessed. Only very high con- centrations of flupirtine antagonized inward currents to NMDA (200 µM) at Ϫ70 mV with an lC against steady-state responses of 182.1 Ϯ 12.1 µM. The
effects of flupirtine were voltage-independent and not associated with recep-tor desensitization making actions within the NMDA receptor channel or atthe glycine modulatory site unlikely. NMDA receptor antagonism probablyhas little relevance for the clinical efficacy of flupirtine as the concentrationsneeded were far higher than those achieved in clinical practice. However, theactivation of a G-protein-regulated inwardly rectifying Kϩ channel wasidentified as an interesting molecular target site of flupirtine. In the next stage,the central nervous spectrum of action of experimental Kϩ channel openers(PCO) was considered. As far as they have been studied, experimental Kϩchannel openers display a spectrum of action comparable to that of flupirtine. In the final stage, a global model was developed in which flupirtine stabilizesthe resting membrane potential by activating inwardly rectifying Kϩ channels,
thus indirectly inhibiting the activation of NMDA receptors. The model pre-sented here reconciles the known functional NMDA receptor antagonism offlupirtine with the activation of Kϩ channels that occurs at therapeutic concen-trations, thus providing an understanding of flupirtine’s spectrum of action. This makes flupirtine the prototype of a clinically applicable substance groupwith analgesic, muscle-relaxant and neuroprotective properties. Keywords: Potassium channel, inwardly rectifying potassium channel, GIRK, flupirtine, N-methyl-D-aspartate receptor antagonist, patch clamp, superior colliculus culture.
Flupirtine (ethyl-N-[2-amino-6-(4-fluorophenylmethylamino)pyridin-3-yl]carbamate) has been in clinical use for many years as a centrally activeanalgesic with muscle-relaxant properties (Friedel and Fitton, 1993). Inpreclinical and preliminary clinical studies, neuroprotective (Schuster et al.,1999), antiepileptic (Sheridan et al., 1986) and antiparkinsonian (Schwarzet al., 1996) effects were additionally found. The mechanism of action offlupirtine has not been clear up to now. Although flupirtine does not haverelevant affinity for any known recognition site on the NMDA receptor com-plex in binding studies (Osborne et al., 1996, 1998), antagonism of this recep-tor has recently been discussed at length as a possible mechanism of action ofthis compound (Osborne et al., 1994; Perovic et al., 1994; Schwarz et al., 1994,1995; Timmann et al., 1995). This assumption is the expression of a misleadingtendency to propose a precise mechanism of action at the receptor levelpurely on the basis of results from behavioral studies or neurotoxicity studiesin vitro with inadequate pharmacological characterization of the interactionsobserved. Such speculations obviously neglect the complexity of neuronalnetworks, interactions between neuronal systems and the importance ofprocesses up- and downstream of receptor activation in mediating end pointparameters such as cell death. In view of this it seemed pertinent to test forpossible interactions between flupirtine and NMDA receptors using wholecell patch clamp recordings from cultured neurones. This approach is bettersuited for the detection of direct antagonists or agonists at any site on theNMDA receptor and thus circumvents the need to propose yet another modu-latory site if no interaction is observed.
Patch clamp recordings from cultured superior colliculus neurones were
performed as described previously (Parsons et al., 1993a). In brief, superiorcolliculi were isolated from embryonic rats (E20–21) and maintained in cul-ture for 11–14 days in NaHCO /HEPES-buffered minimum essential medium
supplemented with 5% foetal calf serum and 5% horse serum (Gibco) andincubated at 37°C with 5% CO (95% humidity). The superior colliculus
culture was chosen for these experiments as it provides very stable recordingconditions which are an absolute prerequisite for voltage-dependency andkinetic experiments. Moreover, the relatively small neurones (soma 15–20 µmØ) are ideally suited to minimise problems of buffered diffusion for concen-tration clamp experiments. Finally, our own unpublished data indicate that
Flupirtine shows functional NMDA receptor antagonism
the somatic NMDA receptors expressed in cultured hippocampal and corticalneurones are similar.
Patch clamp recordings were made from these neurones with polished
glass electrodes (4–5 MΩ) in the whole cell mode at room temperature (20–22°C) with the aid of an EPC-7 amplifier (List). Test substances were appliedby switching channels of a custom made fast superfusion system with a com-mon outflow (Ͻ10 ms exchange times). The contents of the intracellular solu-tion were as follows (mM): CsCl (120), TEACl (20), EGTA (10), MgCl (1),
CaCl (0.2), Glucose (10), ATP (2), cAMP (0.25); pH was adjusted to 7.3 with
CsOH or HCl. The extracellular solutions had the following basic composi-tion (mM): NaCl (140), KCl (3), CaCl (0.2), glucose (10), HEPES (10),
sucrose (4.5), tetrodotoxin (0.3 µM), glycine (1 µM), (pH 7.3). Flupirtine male-ate was a generous gift of G. Pergande, ASTA Medica, Frankfurt. All othercompounds were obtained from Sigma.
Very high concentrations of flupirtine antagonized inward current re-
sponses to NMDA (200 µM) at Ϫ70 mV (Fig. 1A) with an lC against steady-
state responses of 182.1 Ϯ 12.1 µM (Fig. 1B). Flupirtine did not enhanceglycine-dependent desensitization in the continuous presence of the non-saturating concentration of glycine (1 µM), as evidence by the similar potencyagainst the peak component of NMDA-induced currents (IC ϭ 228.6 Ϯ
8.9 µM). This probably excludes antagonistic actions at the strychnine-insensitive glycine modulatory (glycine ) site of the NMDA receptor complex
(Mayer et al., 1989; Parsons et al., 1993b) as previous observations indicatethat most moderate to low affinity full glycine antagonists are three to ten
times more potent against steady-state than against peak currents. The effectsof flupirtine (300 µM) were not voltage-dependent (Fig. 1C) and were appar-ently not use-dependent making channel blockade an unlikely candidate asthe mechanism of NMDA receptor antagonism (Parsons et al., 1995). The Hillcoeff. for NMDA receptor antagonism was close to unity and does not giveany indication for cooperativity. Flupirtine alone did not evoke any measur-able inward or outward current which also excludes direct agonistic actionsat inhibitory GABA or classical glycine receptors as a possible mechanism
of action as both of these receptors are expressed in these cultures and canbe activated by their respective agonists under the conditions used (Parsonset al., 1993a).
The further intention of the present contribution was to provide an under-
standing of the mechanism of action. A four-stage strategy was selected to thisend. The first stage was to determine the therapeutic serum concentration onthe basis of the published data. In a second stage, the in-vitro effects offlupirtine were assessed to determine whether they are relevant under thera-peutic conditions. In the next stage, flupirtine’s spectrum of action wascompared with that of experimental Kϩ channel openers. Finally, a globalmodel was developed to explain the clinical spectrum of action. In this contri-bution, we show that the spectrum of action of flupirtine can be understoodwithin the context of the opening of inwardly rectifying Kϩ channels. Flupirtine is thus the prototype of a new clinically applicable substance groupwith analgesic, muscle-relaxant and neuroprotective properties. Fig. 1. A Concentration-dependence of the blockade of NMDA receptors by flupirtine on a single superior colliculus neurone. NMDA (200 µM) was applied for 2.5 seconds every 30 seconds in the continuous presence of glycine (1 µM) and at a constant mem- brane potential of Ϫ70 mV. The inter-response interval has been omitted to allow better resolution of the kinetics of individual responses (blank spaces in trace). Flupirtine (30, 100 and 300 µM) was continuously present for 1.5 minutes as indicated by the bars. B Concentration-dependence of the blockade of NMDA receptors by flupirtine. Peak and plateau (steady state) NMDA current response were normalised to control levels and plotted as means (ϮSEM) against flupirtine concentration (10 µM n ϭ 3, 30 µM n ϭ 7, 100 µM n ϭ 7, 300 µM n ϭ 5). Estimation of IC s and curve fitting were made according
to the 4 parameter logistic equation (Grafit, Erithacus Software). C Voltage-indepen- dence of the blockade of NMDA receptors by flupirtine. NMDA (200 µM) was applied for 2.5 seconds every 30 seconds in the continuous presence of glycine (1 µM) at various membrane potentials. Plateau NMDA current responses in the absence and presence of flupirtine (300 µM) have been plotted as means against membrane potential (n ϭ 2). The upper left insert shows original data for the i.v. curve in the presence of flupirtine
Flupirtine shows functional NMDA receptor antagonism
Therapeutically relevant concentrations of flupirtine
The therapeutically relevant concentrations of flupirtine are of decisive im-portance for assessing its molecular mechanisms of action. Of the variousexperimentally demonstrated effects of flupirtine (Table 1), only those thatoccur within a concentration range achieved under therapeutic conditions arerelevant. Animal experiments show higher values in the brain than in theplasma 15 min after intravenous administration. After oral administration,comparable concentrations in the plasma and brain are measuredwithin 30 min (Obermeier et al., 1985). Under therapeutic conditions, theplasma concentration is up to 6.5 µM; 2.5 µM are still detectable 12 h afterthe last intake (Hlavica and Niebch, 1985). The elimination half-life of
Table 1. A selection of the in-vitro effects of flupirtine
* Kornhuber et al. (publication in preparation). Under therapeutic conditions with serum concentrations
of up to about 5 µM, of the effects listed here, only the effect on the inwardly rectifying Kϩ channel is relevant
flupirtine is longer in older patients than in young normal subjects; this isaccompanied by higher maximum serum concentrations in older patients(Abrams et al., 1988). The same probably applies for preclinical models wherepronounced effects are seen with doses of around 1–20 mg/kg in vivo (Blocket al., 1994; Carlsson and Jurna, 1987; Schwarz et al., 1994, 1995; Timmann etal., 1995) and 10–20 µM in vitro (Nickel et al., 1990a; Osborne et al., 1994;Perovic et al., 1994; Rupalla et al., 1995). In summary, experimentally deter-mined effects are only clinically relevant if they occur in the low micromolarrange. Pharmacologic effects at therapeutic concentrations
The pain-relieving effect of flupirtine does not appear to be achieved viacentral opiodergic mechanisms. For example, the analgesic effects of flu-pirtine are not antagonized by naloxone, and neither are they accompanied bytolerance or physical dependence of the opiate type (Nickel et al., 1985). Moreover, flupirtine does not show any relevant affinity to the opiate receptorsystem (Nickel, 1987; Nickel et al., 1985) (Table 1) and is also structurallymarkedly different from morphine. An action via benzodiazepine receptorscould also be ruled out (Nickel et al., 1990b). The serotonin receptor an-tagonist cyproheptadine and the tryptophan hydroxylase inhibitor p-chloro-phenylalanine do not inhibit the analgesic properties of flupirtine (Nickelet al., 1985), which also suggests that there is no influence on serotonergicmechanisms. Although no relevant affinities to the α - or α -adrenoreceptors
have been found (Szelenyi et al., 1989), indirect evidence suggests a modula-tion of pain perception via the descending noradrenergic system (Szelenyiet al., 1989).
The central nervous system effects of flupirtine include analgesia, muscle
relaxation and neuroprotection. Antiepileptic and antiparkinsonian proper-ties are also to be found. These properties are only independent of each otherat first sight: they are in fact the typical and classical effects of N-methyl-D-aspartate (NMDA) receptor antagonists (Zieglgänsberger and Tölle, 1993;Kornhuber and Weller, 1997). In various indirect studies, flupirtine displayedproperties that are consistent with antagonism at the NMDA receptor (Blocket al., 1994; Rupalla et al., 1995; Perovic et al., 1994, 1995; Osborne et al., 1994,1996; Schwarz et al., 1994, 1995), and the recent review articles on flupirtinefocus on the NMDA receptor as the main molecular target of flupirtine(Osborne et al., 1998; Schuster et al., 1999). In direct testing, however, it hasnot been possible to demonstrate a clear interaction with the previouslyknown binding sites of the NMDA receptor (Table 1). In patch-clamp inves-tigations on neuronal cell cultures, flupirtine has no influence on NMDA-induced ion currents (results presented here and Jakob and Krieglstein,1997). In our own investigations (Kornhuber et al., publication in prepara-tion), flupirtine did not show any relevant affinity to binding sites at theNMDA receptor in human post-mortem brain tissue. Other research groupshave reported comparable negative results from binding studies (Osborneet al., 1996, 1998). Recently, it has been suggested that flupirtine interacts with
Flupirtine shows functional NMDA receptor antagonism
the redox binding site of the NMDA receptor (Osborne et al., 1998). But thisis inconsistent with the negative findings from patch-clamp studies presentedhere. In summary, it can be stated that flupirtine acts like an NMDA receptorantagonist in functional investigations, although an action at the NMDAreceptor could not be found in direct investigations. It is probable that a siteof action “up- or downstream” of the NMDA receptor is influenced, leadingto a functional NMDA receptor antagonism.
Jakob and Kriegelstein (1997) found an activation of G-protein-regulated
inwardly rectifying Kϩ channels by flupirtine in therapeutically relevantconcentration ranges. According to our current knowledge, this is the onlymechanism known to be relevant in a therapeutic concentration range (Table1). Inwardly rectifying Kϩ channels represent a new family of Kϩ channels anddiffer markedly from the classical voltage-dependent Kϩ channels. The restingmembrane potential is slightly above the Kϩ equilibrium potential; a slightoutflow of Kϩ ions stabilizes the resting membrane potential close to theKϩ equilibrium potential. The overall effect on the cell is a stabilization ofthe resting membrane potential, e.g. in the case of slight depolarization byexcitotoxic stimuli. The G-protein-activated inwardly rectifying Kϩ channels(GIRK) are regulated via neurotransmitters, occur in various subtypes andare expressed differently according to the region of the brain involved(Karschin et al., 1994). Kϩ channels also play an important role in the trans-mission of pain stimuli. The analgesic effects of opioids (Ocaña et al., 1990),
α -adrenergic agonists (Ocaña and Baeyens, 1993), 5-HT Ϫ agonists (Robles
et al., 1996) and other analgetic substances are mediated by receptor-medi-ated opening of Kϩ channels and neuronal hyperpolarization. On the otherhand, Substance P inhibits G-protein-dependent Kϩ channels (Stanfield et al.,1985) and thus facilitates the transmission of pain stimuli. Central nervous effects of flupirtine compared to those of experimental K؉ channel openers
Experimental Kϩ channel openers like cromakalim display analgesic proper-ties (Ocaña et al., 1996; Robles et al., 1996; Vergoni et al., 1992). Directevidence of a neuroprotective action of openers of ligand-gated Kϩ channelshas been found in excitotoxic (Abele and Miller, 1990) and oxidative noxae(Goodman and Mattson, 1996). A study conducted by Lauritzen et al. (1997)shows that cromakalim prevents the glutamate-induced death of hippocampalneurons by counteracting the delayed increase in intracellular Ca2ϩ. Ananalogy to the effects of flupirtine can be seen here (Zimmer et al., 1998). Astabilization of the resting membrane potential by flupirtine would also beconsistent with the initial evidence of antiepileptic properties (Sheridan et al.,1986). Antiepileptic properties have been shown for other Kϩ channelopeners (Gandolfo et al., 1989; Del Pozo et al., 1990; Popoli et al., 1991). Insummary, a comparable central nervous spectrum of action is found forflupirtine and experimental Kϩ channel openers. This can be interpreted asadditional indirect evidence of an action of flupirtine via an activation of Kϩchannels. Global model and summary
The results obtained so far can be summarized as follows: Therapeuticallyrelevant, analgesic plasma concentrations of flupirtine are in the low micro-molar range. In direct investigations, no relevant affinities for α , α , 5HT ,
5HT , dopamine, benzodiazepine, opiate, central muscarinergic, or nicotinic
receptors were found. The profile of preclinical and clinical actions (analgesic,muscle-relaxant, neuroprotective, antiepileptic and antiparkinsonian proper-ties) suggests that the action of flupirtine is connected with the NMDA recep-tor. It has not been possible to convincingly demonstrate a direct action on theNMDA receptor to date. All previous experimental results could be mediatedby an indirect influence on the NMDA receptor. Flupirtine acts functionallylike an NMDA receptor antagonist. At a therapeutically relevant concentra-tion, flupirtine activates neuronal inwardly rectifying G-protein-regulated Kϩchannels. The spectrum of action of the available experimental Kϩ channelopeners, as far as they have been investigated to date, corresponds to that offlupirtine: These Kϩ channel openers also display analgesic, neuroprotectiveand anticonvulsant properties.
We present a new model to explain the spectrum of action of flupirtine:
Flupirtine activates inwardly rectifying Kϩ channels and thus stabilizes theresting membrane potential. The Mg2ϩ block of the NMDA receptor remainsin force; i.e. the NMDA receptor is indirectly inhibited (Fig. 2). This mecha-nism provides an explanation for the analgesia, muscle relaxation andneuroprotection. The model on the mechanism of action of flupirtinepresented here links neuronal Kϩ channels with NMDA receptors via mem-brane excitability. This provides an understanding of the clinically observedprofile of flupirtine’s actions, with analgesic, muscle-relaxant and neuro-protective effects. The indirect inhibition of Ca2ϩ inflow into nerve cells, withplastic changes, e.g. in the sense of an increased response (“wind up”), issuppressed by substances like flupirtine. This counteracts the clinically corre-sponding chronification of pain. From a clinical point of view, flupirtine can beclassified as the prototype of a new substance class with analgesic, muscle-relaxant and neuroprotective properties. While flupirtine is clinically usedmainly as an analgesic substance, its neuroprotective properties will probablylead to new clinical applications, e.g. in chronic neurodegenerative diseases.
Various questions remain open. Different nerve cells often differ in their
intrinsic electrophysiological properties as a result of the differential expres-sion of specific ion channels and their different spatial distribution on the cellsurface. In future investigations, it will have to be clarified which subtype ofGIRK channels is influenced, in which regions of the brain this takes place,and whether specific receptors, G-proteins or Kϩ channels are influenced, andthe precise molecular target site will have to be characterized. Cardiac sideeffects of flupirtine have not been reported to date. This can be interpreted asindirect evidence that flupirtine selectively influences neuronal Kϩ channels. This hypothesis must be examined in further investigations. If this hypothesisis confirmed, flupirtine is the prototype of a new substance class, the selectiveneuronal potassium channel openers (SNEPCO).
Flupirtine shows functional NMDA receptor antagonism
Fig. 2. Flupirtine activates G-protein-coupled inwardly rectifying Kϩ channels. The changes under the influence of flupirtine are to be read from left to right. The resting membrane potential is stabilized, an activation of the cell membrane is inhibited. These processes are shown here in different grey tones of the cell membrane: The activated state of the cell membrane (left, dark) is brought into the resting state (right, light) by flupirtine via an activation of inwardly rectifying Kϩ channels. An activation of NMDA receptors is prevented, since the Mg2ϩ block of the NMDA receptor is only relieved upon depolariza- tion of the cell membrane. It is conceivable that flupirtine has additional effects that are Acknowledgements
This study was supported by the German Ministry of Education, Science and Technology(JK, 01 EB 9410). References
Abele AE, Miller RJ (1990) Potassium channel activators abolish excitotoxicity in cul-
tured hippocampal pyramidal neurons. Neurosci Lett 115: 195–200
Abrams SML, Baker LRI, Crome P, White AST, Johnston A, Ankier SI, Warrington SJ,
Turner P, Niebch G (1988) Pharmacokinetics of flupirtine in elderly volunteers and inpatients with moderate renal impairment. Postgrad Med J 64: 361–363
Block F, Pergande G, Schwarz M (1994) Flupirtine protects against ischaemic retinal
dysfunction in rats. NeuroReport 5: 2630–2632
Carlsson KH, Jurna I (1987) Depression by flupirtine, a novel analgesic agent, of motor
and sensory responses of the nociceptive system in the rat spinal cord. Eur JPharmacol 143: 89–99
Darius H, Schrör K (1985) The action of flupirtine on prostaglandin formation and
platelet aggregation in vitro. Arzneimittelforschung 35: 55–59
Del Pozo E, Barrios M, Baeyens JM (1990) Effects of potassium channel openers on
pentylenetetrazole-induced seizures in mice. Pharmacol Toxicol 67: 182–184
Friedel HA, Fitton A (1993) Flupirtine: a review of its pharmacological properties and
therapeutic efficacy in pain states. Drugs 45: 548–569
Gandolfo G, Romettino S, Gottesmann C, van Luijtelaar G, Coenen A, Bidard JN,
Lazdunski M (1989) Kϩ channel openers decrease seizures in genetically epilepticrats. Eur J Pharmacol 167: 181–183
Goodman Y, Mattson MP (1996) Kϩ channel openers protect hippocampal neu-
rons against oxidative injury and amyloid -peptide toxicity. Brain Res 706: 328–332
Hlavica P, Niebch G (1985) Untersuchungen zur Pharmakokinetik und Biotransforma-
tion des Analgetikums Flupirtin beim Menschen. Arzneimittelforschung 35: 67–74
Jakob R, Krieglstein J (1997) Influence of flupirtine on a G-protein coupled inwardly
rectifying potassium current in hippocampal neurones. Br J Pharmacol 122: 1333–1338
Karschin C, Schreibmayer W, Dascal N, Lester H, Davidson N, Karschin A (1994)
Distribution and localization of a G protein-coupled inwardly rectifying Kϩ channelin the rat. FEBS Lett 348: 139–144
Kornhuber J, Weller M (1997) Psychotogenicity and N-methyl-D-aspartate receptor
antagonism: implications for neuroprotective pharmacotherapy. Biol Psychiatry 41:135–144
Lauritzen I, De Weille JR, Lazdunski M (1997) The potassium channel opener (—)-
cromakalim prevents glutamate-induced cell death in hippocampal neurons. JNeurochem 69: 1570–1579
Mayer ML, Vyklicky L, Clements J (1989) Regulation of NMDA receptor desensitization
in mouse hippocampal neurons by glycine. Nature 338: 425–427
Nickel B (1987) The antinociceptive activity of flupirtine: a structurally new analgesic.
Nickel B, Herz A, Jakovlev V, Tiebes U (1985) Untersuchungen zum Wirkungsme-
chanismus des Analgetikums Flupirtin. Arzneimittelforschung 35: 1402–1409
Nickel B, Jakovlev V, Szelenyi I (1990a) Einfluß von Flupirtin, verschiedener Analgetika
und Muskelrelaxantien auf den Skelettmuskeltonus wacher Ratten. Arzneimittel-forschung 40: 909–911
Nickel B, Borbe HO, Szelenyi I (1990b) Investigations with the novel non-opioid anal-
gesic flupirtine in regard to possible benzodiazepine-like abuse inducing potential. Arzneimittelforschung 40: 905–908
Obermeier K, Niebch G, Thiemer K (1985) Untersuchungen zur Pharmakokinetik und
Biotransformation des Analgetikums Flupirtin bei Ratte und Hund. Arzneimittel-forschung 35: 60–67
Ocaña M, Baeyens JM (1993) Differential effects of Kϩ channel blockers on anti-
nociception induced by α -adrenoceptor, GABA and k-opioid receptor agonists. Br
Ocaña M, Del Pozo E, Barrios M, Robles LI, Baeyens JM (1990) An ATP-dependent
potassium channel blocker antagonizes morphine analgesia. Eur J Pharmacol 186:377–378
Ocaña M, Barrios M, Baeyens JM (1996) Cromakalim differentially enhances
antinociception induced by agonists of alpha adrenoceptors, γ-aminobutyric acid ,
mu and kappa opioid receptors. J Pharmacol Exp Ther 276: 1136–1142
Osborne NN, Pergande G, Block F, Schwarz M (1994) Immunohistochemical evidence
for flupirtine acting as an antagonist on the N-methyl-D-aspartate and homocysteicacid-induced release of GABA in the rabbit retina. Brain Res 667: 291–294
Osborne NN, Schwarz M, Pergande G (1996) Protection of rabbit retina from ischemic
injury by flupirtine. Invest Ophthalmol Vis Sci 37: 274–280
Osborne NN, Cazevieille C, Wood JPM, Nash MS, Pergande G, Block F, Kosinski C,
Schwarz M (1998) Flupirtine, a nonopioid centrally acting analgesic, acts as anNMDA antagonist. Gen Pharmacol 30: 255–263
Parsons CG, Gruner R, Rozental J, Millar J, Lodge D (1993a) Patch clamp studies on the
kinetic and selectivity of N-methyl-D-aspartate receptor antagonism by memantine(1-amino-3,5-dimethyladamantan). Neuropharmacology 32: 1337–1350
Parsons CG, Zong X, Lux HD (1993b) Whole cell and single channel analysis of the
kinetics of glycine-sensitive N-methyl-D-aspartate receptor desensitization. Br JPharmacol 109: 213–221
Flupirtine shows functional NMDA receptor antagonism
Parsons CG, Quack G, Bresink I, Baran L, Przegalinski E, Kostowski W, Krzascik P,
Hartmann S, Danysz W (1995) Comparison of the potency, kinetics andvoltage-dependency of open channel blockade for a series of uncompetitive NMDAantagonists in vitro with anticonvulsive and motor impairment activity in vivo. Neu-ropharmacology 34: 1239–1258
Perovic S, Schleger C, Pergande G, Iskric I, Ushijima H, Rytic P, Müller WEG (1994) The
triaminopyridine flupirtine prevents cell death in rat cortical cells induced by N-methyl-D-aspartate and gp 120 of HIV-1. Eur J Pharmacol Mol Pharmacol Sect 288:27–33
Perovic S, Pergande G, Ushijima H, Kelve M, Forrest J, Müller WEG (1995) Flupirtine
partially prevents neuronal injury induced by prion protein fragment and leadacetate. Neurodegeneration 4: 369–374
Popoli P, Pezzola A, Sagratella S, Zeng YC, Scotti de Carolis A (1991) Cromakalim (BRL
34915) counteracts the epileptiform activity elicited by diltiazem and verapamil inrats. Br J Pharmacol 104: 907–913
Robles LI, Barrios M, Del Pozo E, Dordal A, Baeyens JM (1996) Effects of Kϩ channel
blockers and openers on antinociception induced by agonists of 5-HT receptors.
Rupalla K, Cao W, Krieglstein J (1995) Flupirtine protects neurons against excitotoxic or
ischemic damage and inhibits the increase in cytosolic Ca2ϩ concentration. Eur JPharmacol 294: 469–473
Schuster G, Schwarz M, Block F, Pergande G, Schmidt WJ (1999) Flupirtine: a review of
its neuroprotective and behavioral properties. CNS Drugs (in press)
Schwarz M, Block F, Pergande G (1994) N-methyl-D-aspartate (NMDA)-mediated
muscle relaxant action of flupirtine in rats. NeuroReport 5: 1981–1984
Schwarz M, Schmitt T, Pergande G, Block F (1995) N-methyl-D-aspartate and α -
adrenergic mechanisms are involved in the depressant action of flupirtine on spinalreflexes in rats. Eur J Pharmacol 276: 247–255
Schwarz M, Nolden-Koch M, Purr J, Pergande G, Block F (1996) Antiparkinsonian effect
of flupirtine in monoamine-depleted rats. J Neural Transm 103: 581–590
Sheridan PH, Seaman CA, Narang PK, White BG, Schwerdt P, Theodore WH, Rose DF,
Nice FJ, Gallelli JF, Mirsky AF, Engel J, Porter RJ (1986) Pilot study of flupirtine inrefractory seizures. Neurology 36 [Suppl 1]: 85
Stanfield PR, Nakajima Y, Yamaguchi K (1985) Substance P raises neuronal membrane
excitability by reducing inward rectification. Nature 315: 498–501
Szelenyi I, Nickel B, Borbe HO, Brune K (1989) Mode of antinociceptive action of
flupirtine in the rat. Br J Pharmacol 97: 835–842
Timmann D, Plummer C, Schwarz M, Diener HC (1995) Influence of flupirtine on human
lower limb reflexes. Electroencephalogr Clin Neurophysiol 97: 184–188
Vergoni AV, Scarano A, Bertolini A (1992) Pinacidil potentiates morphine analgesia.
Zieglgänsberger W, Tölle TR (1993) The pharmacology of pain signalling. Curr Opin
Zimmer G, Balakirev M, Zwicker K, Hofmann M, Woodcock BG, Pergande G (1998)
Effect of the triaminopyridine flupirtine on calcium uptake, membrane potential andATP synthesis in rat heart mitochondria. Br J Pharmacol 123: 1154–1158
Authors’ address: Prof. Dr. J. Kornhuber, Abteilung für Psychiatrie, Universität
Göttingen, von-Siebold-Strasse 5, D-37075 Göttingen, Federal Republic of Germany,e-mail: jkornhu@gwdg.de
Persian Journal of Acarology, Vol. 2, No. 2, pp. 335–339. Corresspondence Mites associated with the date palm ( Phoenix dactylifera L.) in Larestan (Fars province), southern Iran Department of Plant Protection, Faculty of Agriculture, Shiraz University, Shiraz, Iran; e-mails: maryam_majidi81@ yahoo.com; akrami@shirazu.ac.ir Date palm ( Phoenix dactylifera L.) is one of t
Tribunal d'appel en Tribunal matière de permis APPEAL UNDER SECTION 50(1) OF THE HIGHWAY TRAFFIC ACT , R.S.O. 1990, c. H.8, FROM A DECISION OF THE REGISTRAR OF MOTOR VEHICLES PURSUANT TO SECTION 47(1) OF THAT ACT CARMICHAEL, Agent representing the Applicant KYLE BIEL, Agent representing the Registrar of Motor Vehicles DECISION AND REASONS This is an appeal to the Licence
 Flupirtine shows functional NMDA receptor antagonism by
Flupirtine shows functional NMDA receptor antagonism by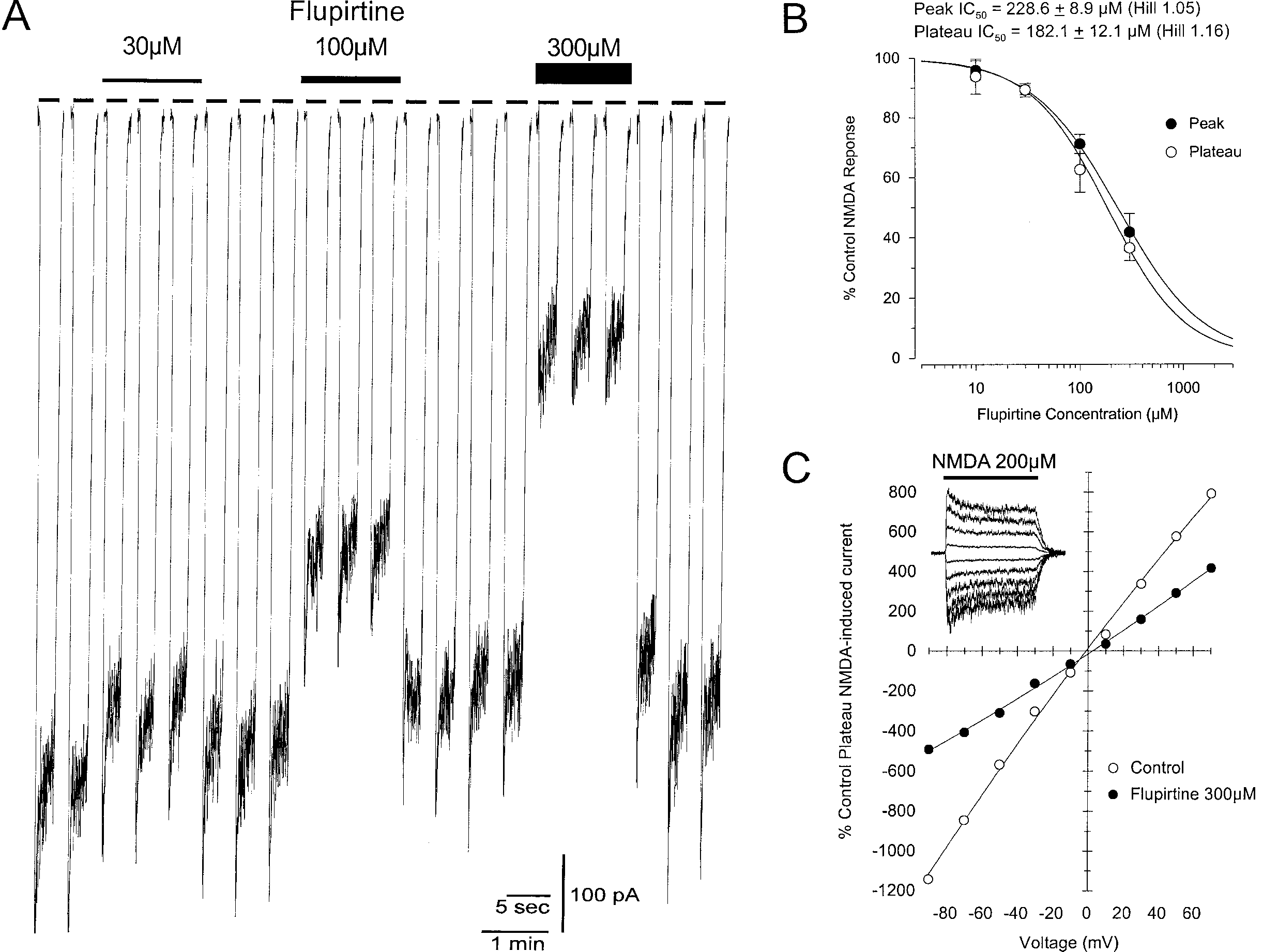 Fig. 1. A Concentration-dependence of the blockade of NMDA receptors by flupirtine
Fig. 1. A Concentration-dependence of the blockade of NMDA receptors by flupirtine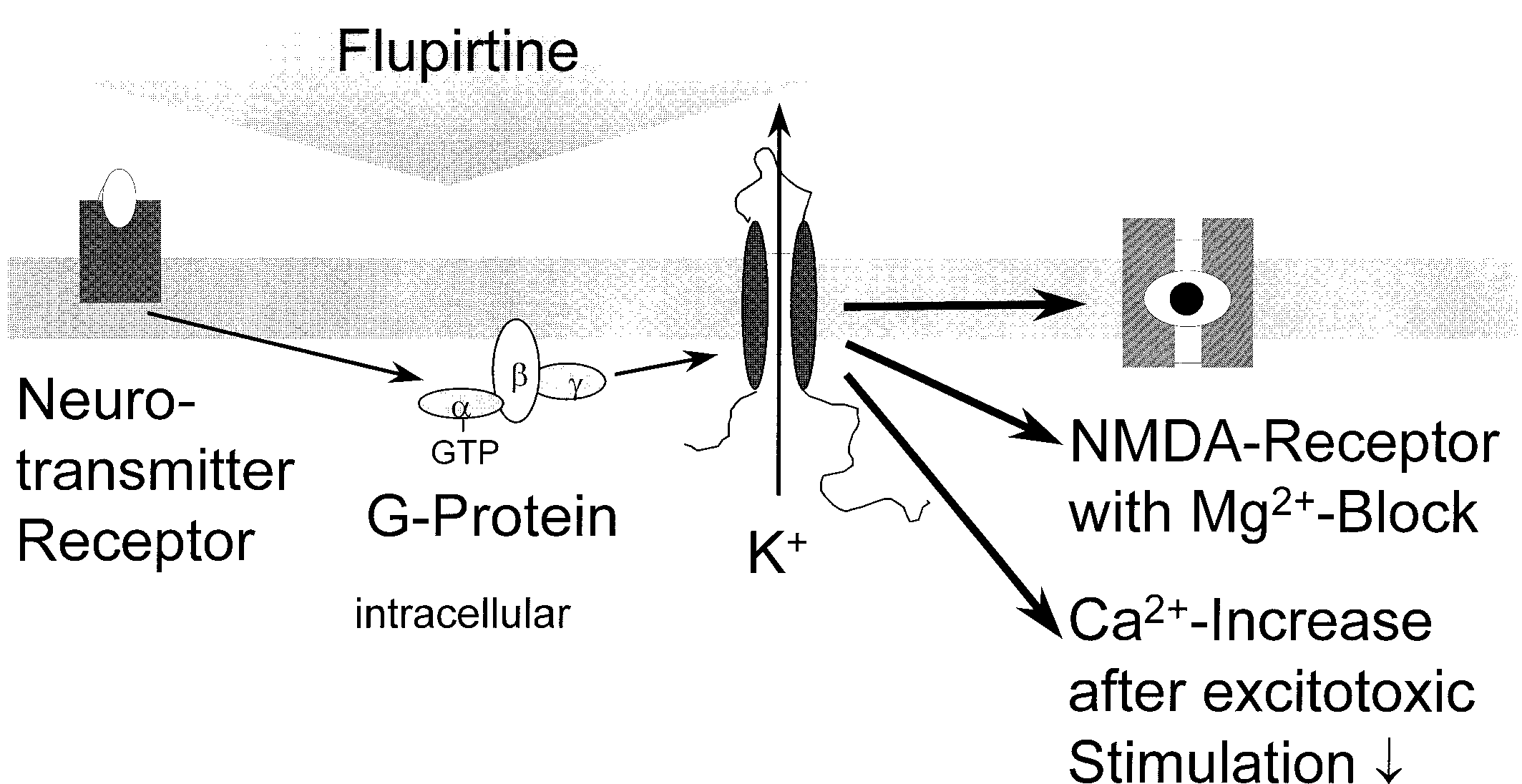 Flupirtine shows functional NMDA receptor antagonism
Fig. 2. Flupirtine activates G-protein-coupled inwardly rectifying Kϩ channels. The
Flupirtine shows functional NMDA receptor antagonism
Fig. 2. Flupirtine activates G-protein-coupled inwardly rectifying Kϩ channels. The